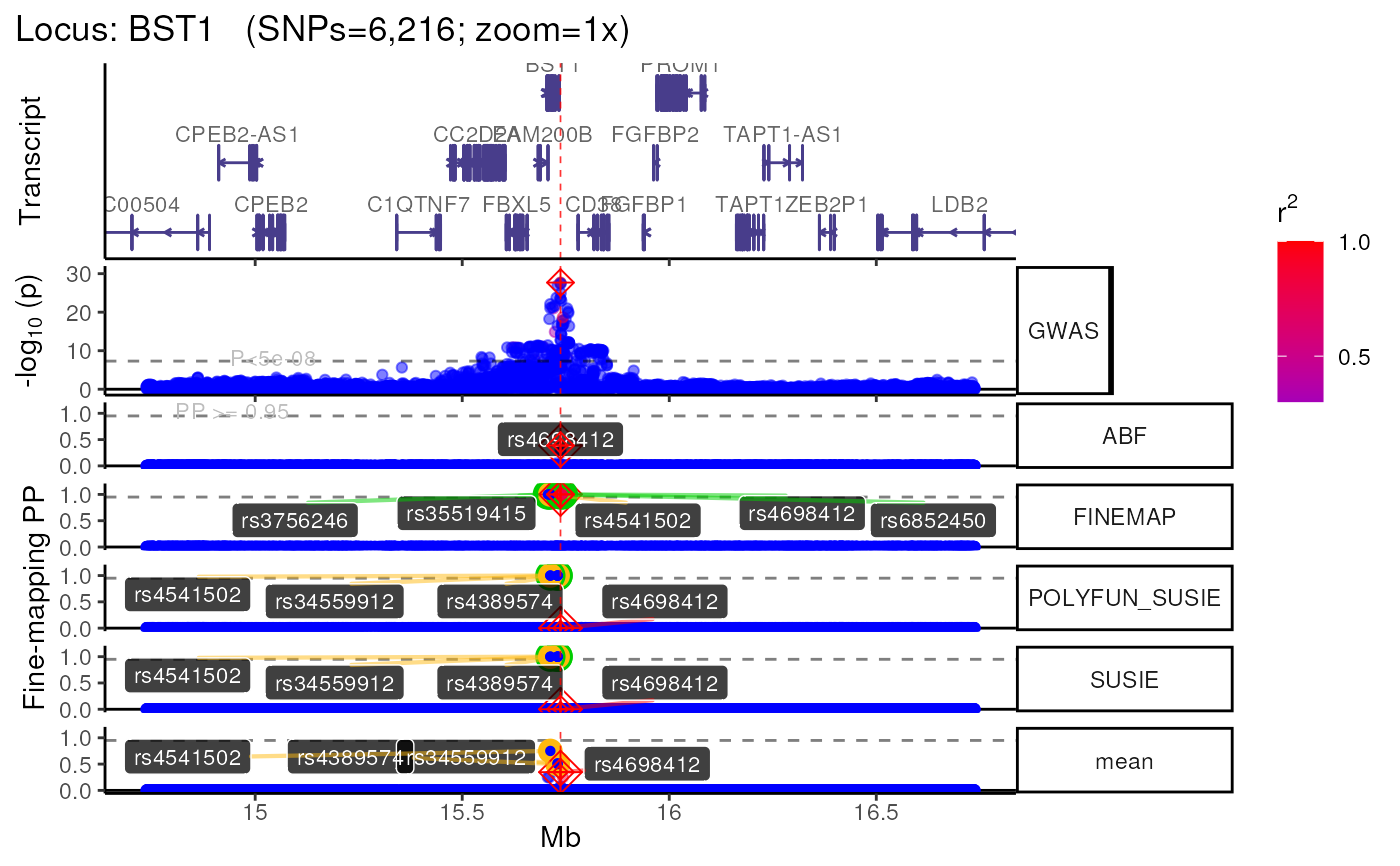
Generate a locus-specific plot with multiple selectable tracks. Users can also generate multiple zoomed in views of the plot at multiple resolutions.
Usage
plot_locus(
dat,
locus_dir,
LD_matrix = NULL,
LD_reference = NULL,
facet_formula = "Method~.",
dataset_type = "GWAS",
color_r2 = TRUE,
finemap_methods = c("ABF", "FINEMAP", "SUSIE", "POLYFUN_SUSIE"),
track_order = NULL,
track_heights = NULL,
plot_full_window = TRUE,
dot_summary = FALSE,
qtl_suffixes = NULL,
mean.PP = TRUE,
credset_thresh = 0.95,
consensus_thresh = 2,
sig_cutoff = 5e-08,
gene_track = TRUE,
tx_biotypes = NULL,
point_size = 1,
point_alpha = 0.6,
density_adjust = 0.2,
snp_group_lines = c("Lead", "UCS", "Consensus"),
labels_subset = c("Lead", "CS", "Consensus"),
xtext = FALSE,
show_legend_genes = TRUE,
xgr_libnames = NULL,
xgr_n_top = 5,
roadmap = FALSE,
roadmap_query = NULL,
roadmap_n_top = 7,
zoom_exceptions_str = "*full window$|zoom_polygon",
nott_epigenome = FALSE,
nott_regulatory_rects = TRUE,
nott_show_placseq = TRUE,
nott_binwidth = 200,
nott_bigwig_dir = NULL,
save_plot = FALSE,
show_plot = TRUE,
genomic_units = "Mb",
strip.text.y.angle = 0,
max_transcripts = 1,
zoom = c("1x"),
dpi = 300,
height = 12,
width = 10,
plot_format = "png",
save_RDS = FALSE,
return_list = FALSE,
conda_env = "echoR_mini",
nThread = 1,
verbose = TRUE
)Arguments
- dat
Data to query transcripts with.
- locus_dir
Storage directory to use.
- LD_matrix
LD matrix.
- LD_reference
LD reference to use:
- 1KGphase1
1000 Genomes Project Phase 1 (genome build: hg19).
- 1KGphase3
1000 Genomes Project Phase 3 (genome build: hg19).
- UKB
Pre-computed LD from a British European-decent subset of UK Biobank. Genome build : hg19
- <vcf_path>
User-supplied path to a custom VCF file to compute LD matrix from.
Accepted formats: .vcf / .vcf.gz / .vcf.bgz
Genome build : defined by user withtarget_genome.- <matrix_path>
User-supplied path to a pre-computed LD matrix. Accepted formats: .rds / .rda / .csv / .tsv / .txt
Genome build : defined by user withtarget_genome.
- facet_formula
Formula to facet plots by. See facet_grid for details.
- dataset_type
Dataset type (e.g. "GWAS" or "eQTL").
- color_r2
Whether to color data points (SNPs) by how strongly they correlate with the lead SNP (i.g. LD measured in terms of r2).
- finemap_methods
Fine-mapping methods to plot tracks for, where the y-axis show the Posterior Probabilities (PP) of each SNP being causal.
- track_order
The order in which tracks should appear (from top to bottom).
- track_heights
The height of each track (from top to bottom).
- plot_full_window
Include a track with a Manhattan plot of the full GWAS/eQTL locus (not just the zoomed-in portion).
- dot_summary
Include a dot-summary plot that highlights the Lead, Credible Set, and Consensus SNPs.
- qtl_suffixes
If columns with QTL data is included in
dat, you can indicate which columns those are with one or more string suffixes (e.g.qtl_suffixes=c(".eQTL1",".eQTL2")to use the columns "P.QTL1", "Effect.QTL1", "P.QTL2", "Effect.QTL2").- mean.PP
Include a track showing mean Posterior Probabilities (PP) averaged across all fine-mapping methods.
- credset_thresh
The minimum fine-mapped posterior probability for a SNP to be considered part of a Credible Set. For example,
credset_thresh=.95means that all Credible Set SNPs will be 95% Credible Set SNPs.- consensus_thresh
The minimum number of fine-mapping tools in which a SNP is in the Credible Set in order to be included in the "Consensus_SNP" column.
- sig_cutoff
Filters out SNPs to plot based on an (uncorrected) p-value significance cutoff.
- gene_track
Include a track showing gene bodies.
- tx_biotypes
Transcript biotypes to include in the gene model track. By default (
NULL), all transcript biotypes will be included. See get_tx_biotypes for a full list of all available biotypes- point_size
Size of each data point.
- point_alpha
Opacity of each data point.
- density_adjust
Passed to
adjustargument in geom_density.- snp_group_lines
Include vertical lines to help highlight SNPs belonging to one or more of the following groups: Lead, Credible Set, Consensus.
- labels_subset
Include colored shapes and RSID labels to help highlight SNPs belonging to one or more of the following groups: Lead, Credible Set, Consensus.
- xtext
Include x-axis title and text for each track (not just the lower-most one).
- show_legend_genes
Show the legend for the
gene_track.- xgr_libnames
Passed to XGR_plot. Which XGR annotations to check overlap with. For full list of libraries see here (XGR on CRAN). Passed to the
RData.customisedargument inXGR::xRDataLoader. Examples:"ENCODE_TFBS_ClusteredV3_CellTypes""ENCODE_DNaseI_ClusteredV3_CellTypes""Broad_Histone"
- xgr_n_top
Passed to XGR_plot. Number of top annotations to be plotted (passed to XGR_filter_sources and then XGR_filter_assays).
- roadmap
Find and plot annotations from Roadmap.
- roadmap_query
Only plot annotations from Roadmap whose metadata contains a string or any items from a list of strings (e.g.
"brain"orc("brain","liver","monocytes")).- roadmap_n_top
Passed to ROADMAP_plot. Number of top annotations to be plotted (passed to ROADMAP_query).
- zoom_exceptions_str
Names of tracks to exclude when zooming.
- nott_epigenome
Include tracks showing brain cell-type-specific epigenomic data from Nott et al. (2019) (doi:10.1126/science.aay0793 ).
- nott_regulatory_rects
Include track generated by NOTT2019_epigenomic_histograms.
- nott_show_placseq
Include track generated by NOTT2019_plac_seq_plot.
- nott_binwidth
When including Nott et al. (2019) epigenomic data in the track plots, adjust the bin width of the histograms.
- nott_bigwig_dir
Instead of pulling Nott et al. (2019) epigenomic data from the UCSC Genome Browser, use a set of local bigwig files.
- save_plot
Save plot as RDS file.
- show_plot
Print plot to screen.
- genomic_units
Which genomic units to return window limits in.
- strip.text.y.angle
Angle of the y-axis facet labels.
- max_transcripts
Maximum number of transcripts per gene.
- zoom
Zoom into the center of the locus when plotting (without editing the fine-mapping results file). You can provide either:
The size of your plot window in terms of basepairs (e.g.
zoom=50000for a 50kb window).How much you want to zoom in (e.g.
zoom="1x"for the full locus,zoom="2x"for 2x zoom into the center of the locus, etc.).
You can pass a list of window sizes (e.g.
c(50000,100000,500000)) to automatically generate multiple views of each locus. This can even be a mix of different style inputs: e.g.c("1x","4.5x",25000).- dpi
dpi to use for raster graphics
- height
height (defaults to the height of current plotting window)
- width
width (defaults to the width of current plotting window)
- plot_format
Format to save plot as when saving with ggsave.
- save_RDS
Save the tracks as an RDS file (Warning: These plots take up a lot disk space).
- return_list
Return a named list with each track as a separate plot (default:
FALSE). IfTRUE, will return a merged plot using wrap_plots.- conda_env
Conda environment to use.
- nThread
Number of threads to parallelise across (when applicable).
- verbose
Print messages.
Examples
dat1 <- echodata::BST1
LD_matrix <- echodata::BST1_LD_matrix
locus_dir <- file.path(tempdir(),echodata::locus_dir)
plt <- echoplot::plot_locus(dat = dat1,
locus_dir = locus_dir,
LD_matrix = LD_matrix,
show_plot = TRUE)
#> +-------- Locus Plot: BST1 --------+
#> + support_thresh = 2
#> + Calculating mean Posterior Probability (mean.PP)...
#> + 4 fine-mapping methods used.
#> + 7 Credible Set SNPs identified.
#> + 3 Consensus SNPs identified.
#> + Filling NAs in CS cols with 0.
#> + Filling NAs in PP cols with 0.
#> LD_matrix detected. Coloring SNPs by LD with lead SNP.
#> Filling r/r2 NAs with 0
#> ++ echoplot:: GWAS full window track
#> ++ echoplot:: GWAS track
#> ++ echoplot:: Merged fine-mapping track
#> Melting PP and CS from 5 fine-mapping methods.
#> + echoplot:: Constructing SNP labels.
#> Data is already melted. Skipping.
#> Loading required namespace: ggrepel
#> Adding SNP group labels to locus plot.
#> ++ echoplot:: Adding Gene model track.
#> Converting dat to GRanges object.
#> max_transcripts= 1 .
#> 16 transcripts from 16 genes returned.
#> Fetching data...
#> OK
#> Parsing exons...
#> OK
#> Defining introns...
#> OK
#> Defining UTRs...
#> OK
#> Defining CDS...
#> OK
#> aggregating...
#> Done
#> Constructing graphics...
#> + Adding vertical lines to highlight SNP groups.
#> +>+>+>+>+ zoom = 1x +<+<+<+<+
#> + echoplot:: Get window suffix...
#> + echoplot:: Removing GWAS full window track @ zoom=1x
#> + Removing subplot margins...
#> + Reordering tracks...
#> + Ensuring last track shows genomic units.
#> + Aligning xlimits for each subplot...
#> + Checking track heights...